


Mitochondria are maternally inherited ubiquitous cellular organelles located in the cytoplasm of most eukaryotic cells, with the significant exception of erythrocytes (Gorman et al, 2016). Colloquially referred to as the powerhouse of the cell, mitochondria are pivotal in the synthesis of adenosine triphosphate (ATP), as the tricarboxylic acid cycle (or Krebs cycle) and oxidative phosphorylation (or respiratory chain) occur within this organelle, as well as being involved in a plethora of other fundamental cellular metabolic pathways, including fatty acid oxidation, the urea cycle, gluconeogenesis and ketogenesis (Duchen, 2004; Falzone, 2013; Gorman et al, 2016).
Mitochondrial diseases are a type of metabolic disorder, involving the respiratory chain, under the control of both the mitochondrial DNA (mtDNA) and nuclear DNA (nDNA). When they affect the central nervous system, mitocondriopathies can also be clinically classified as neurodegenerative diseases, alongside myelin disorders (demyelination and leukodystrophies), spongiform degeneration and lysosomal storage diseases (Sisó et al, 2006).
Mitochondrial diseases are increasingly being recognised in human medicine, with clinical presentations being clustered into syndromes and diagnosis relying more on molecular characteristics (for instance with immunohistochemistry or specific genetic testing), rather than on neuropathological hallmarks (DiMauro et al, 2013; Gorman et al, 2016; Davison and Rahman, 2017). In dogs, only a few mitochondriopathies have had their structural, biochemical and genetic basis identified, and in most cases, diagnosis relies on histopathological findings (Vandevelde et al, 2012).
In this review, the authors summarise the basis for suspecting a mitochondrial disease clinically, reviewing the presentations of suspected or confirmed mitochondriopathies in dogs, with an emphasis on mitochondrial encephalopathies, encephalomyelopathies and neuropathies.
Aetiology
The aetiology of mitochondrial diseases classically involves inheritable genetic mutations in mtDNA and nDNA coding for mitochondrial components, as well as de novo mutations within the individual (DiMauro et al, 2013; Gorman et al, 2016). In addition to genetic causes, environmental factors could also play a role in the onset and development of other non-classic mitochondrial disorders (Vernau et al, 2015).
Contributing to the variable clinical presentation in mitochondriopathies, clinical presentation varies because of heteroplasmy, a term describing the presence of two or more variants (mutated and wild-type) mtDNA genomes within an individual cell often caused by de novo mutations (Duchen, 2004; DiMauro et al, 2013; Gorman et al, 2016). Heteroplasmy levels (low or high levels of mutated mtDNA) can also vary between different somatic tissues within the same individual, and occasionally, only specific cells, tissues, or organs become affected by mitochondrial dysfunction (Huang, 2020). All this considered, signs of dysfunction can involve multiple organ systems, although more metabolically active tissues with higher energy requirements, such as the nervous system, heart and skeletal muscle, are commonly affected (Falzone, 2013; Davison and Rahman, 2017).
Epidemiology
In people, childhood-onset mitochondrial diseases are also considered rare, with a reported prevalence of 5–15 cases per 100 000 (Gorman et al, 2016), despite mtDNA disorders being reported to affect around 1 case in 5000 in the overall population (Gorman et al, 2015). Reports of mitochondrial disorders in dogs are still sparse and have traditionally been identified as myopathies, characterised by the presence of ragged red fibres, defined by accumulations of mitochondria in the subsarcolemmal region of muscle fibres, which appear red following staining with modified Gömüri trichrome (Herrtage and Houlton, 1979; Houlton and Herrtage, 1980; Breitschwerdt et al, 1992; Olby et al, 1997; Pacciello, 2003; Tauro et al, 2008). However, the increased availability of diagnostic tools in veterinary medicine have permitted the diagnosis of an expanding number of mitochondrial encephalomyelopathies.
Clinical suspicion and diagnosis
Clinical suspicion of a mitochondrial disease can be raised in the presence of a progressive history of neurological dysfunction, most commonly in young dogs, particularly in the face of a multisystemic disorder. In people, any combination of disease affecting three or more organ systems should prompt consideration of a metabolic pathology, including a mitochondrial disease (Davidson and Rahman et al, 2017). The diagnosis of a mitochondropathy relies on the exclusion of other metabolic diseases and can typically only be confirmed histopathologically. Differential diagnoses include hepatic or renal encephalopathy, organic acidurias, lysosomal storage diseases, nutritional disorders and electrolyte imbalances.
Neurological signs relating to mitochondrial disorders are extremely variable, and only a few syndromes have been recognised in dogs. Clinical phenotypes of mitochondriopathies can involve not only the brain, but also the spinal cord and the peripheral nerves. Despite the variable possible phenotypes, possible ‘red flags’ for a mitochondrial disorder include the association of muscular and neurological clinical signs with concurrent multi-organ involvement, evidence of a progressive or a waxing-and-waning history with subsequent involvement of new body systems (Davison and Rahman, 2017).
Onset of multifocal neurological dysfunction can occur at any age. In people, onset of mitochondrial disease is considered bimodal, with a peak in the first 3 years of life and a second peak from the end of adolescence into the fourth decade of life. In dogs, the vast majority of cases start developing clinical signs early in life, before 1 year of age (Table 1).
Table 1. Syndromes of confirmed or suspected mitochondrial origin in dogs
| Mitochondrial diseases in dogs with a published genetic mutation | Affected breeds and age of onset (AO) | Genetic mutation |
|---|---|---|
| Mainly affecting the central and peripheral nervous system | ||
| Alaskan husky encephalopathy (AHE)/Subacute necrotising encephalomyelopathy (Wakshlag et al, 1999; Brenner et al, 2000; Vernau et al, 2013, 2015) | Alaskan HuskyAO: Before 1 year of age in most cases | Mutation in the thiamine transporter 2 (SLC19A3) gene (Vernau et al, 2013) |
| Leigh-Like subacute necrotising encephalopathy (SNE) (Baiker et al, 2009; Drögemüller et al, 2020) (Figure 1) | Yorkshire TerrierAO: Between 4 and 18 months in most cases, but has been reported up to 5 years | Mutation in the thiamine transporter 2 (SLC19A3) gene (Drögemüller et al, 2020) |
| Inherited spongiform leukoencephalomyelopathy (SLE) (Wood and Patterson, 2001; Li et al, 2006) | Australian Cattle Dog and Shetland SheepdogAO: Most cases at 3–4 weeks of age | Missense mutation in mtDNA encoded cytochrome b (G14474A) (Li et al, 2006) |
| Sensory ataxic neuropathy (SAN) (Jäderlund et al, 2007; Baranowska et al, 2009) | Golden RetrieverAO: 2–8 months of age | Deletion in the mitochondrial tRNATyr gene (Baranowska et al, 2009) |
| Paroxysmal Exercise-Induced Dyskinesia (Nessler et al, 2020) | Shetland Sheepdogs (all female)AO: 2–6 years of age | PCK2 gene encoding the mitochondrial phosphoenolpyruvate carboxykinase 2 (Nessler et al, 2020) |
| Mainly affecting the muscular system | ||
| Mitochondrial myopathy (Griffiths and Duncan, 1979; Herrtage and Houlton, 1979; Houlton and Herrtage, 1980; Shelton et al, 2000; Abramson et al, 2004; Cameron et al, 2007) | Sussex Spaniel/Clumber Spaniel (Cameron et al, 2007)German Shepherd Dog (Paciello et al, 2003), Springer Spaniel (Tauro et al, 2015), Jack Russel Terrier (Olby et al, 1997), Old English Sheepdog (Breitschwerdt et al, 1992; Vijayasarathy et al, 1994), Irish Terrier (Wentink et al, 2010)AO: not reviewed | Pyruvate dehydrogenase deficiency (PDP1) - Clumber Spaniel and Sussex Spaniel (Cameron et al, 2007)Not reported in other breeds |
| Presumed mitochondrial diseases in dogs without a published genetic mutation | ||
| Mainly affecting the central and peripheral nervous system | ||
| Spongiform leucoencephalomyelopathy (SLE) (Martin-Vaquero et al, 2012; Gutierrez-Quintana et al, 2019) | Border TerrierAO: Reported to develop from 3 weeks of age | Genetic test available, but gene involved has not yet been published (University of Missouri) |
| Hereditary polioencephalomyelopathy (Brenner et al, 1997b; Harkin et al, 1999) | Australian Cattle DogAO: 5–24 months of age | Not reported |
| Mitochondriopathy with Regional Encephalic Mineralisation (Gruber et al, 2002) | Jack Russell TerrierAO: 10-month-old dog | Not reported |
| Encephalomyelopathy with morphological abnormalities in mitochondria (Brenner et al, 1997a) | English Springer SpanielAO: 16-month-old dog | Not reported |
| Polioencephalomyelopathy (Kent et al, 2009) | Shih TzuAO: 17-month-old dog | Not reported |
| Polioencephalomyelopathy resembling Leigh's disease (Chai et al, 2015) | CrossbreedAO: 14-month-old dog | Not reported |
| Subacute necrotising encephalopathy (Collins et al, 2013) | American Staffordshire Bull TerrierAO: 6–8 weeks | Not reported |
Haematology, serum biochemistry and urinalysis
Routine haematology and urinalysis are typically within normal limits. Serum biochemistry could raise suspicion of a mitochondrial disease, when increased concentrations of aspartate transaminase (AST), lactate, pyruvate and creatinine kinase (CK) concentrations are found, particularly in myopathies. Located within the hepatocyte's cytosol and mitochondria, AST can be increased in canine mitochondrial myopathies (Paciello et al, 2003; Tauro et al, 2008). Lactate, an end-product of anaerobic metabolism, is found to be increased in states of hypoperfusion and hypoxia, but hyperlactatemia and lactic acidosis can also be found in situations of normal oxygen delivery, namely, secondary to canine mitochondrial myopathies (Beer, 2017). Considering that the respiratory chain occurs within the mitochondria, lactate can be a crude but valuable indication of its correct function. Plasma lactate can be evaluated by measuring its concentrations at rest and following exercise, and measuring the lactate:pyruvate ratio could support claims of a hyperlactatemia (Platt and Garosi, 2004; Collins et al, 2013). Quantitative urine organic acid analysis can reveal elevated lactate and associated metabolites, any abnormal metabolites could be an indication of an organic aciduria, an important differential diagnosis of mitochondrial disorders (Davison and Rahman, 2017).
Cerebrospinal fluid
Cerebrospinal fluid (CSF) total nucleated cell count and protein concentrations are likely to be within normal limits, although CSF analysis of organic acids and amino acids might reveal elevated concentrations of lactate, pyruvate, 3-OH butyric acid, and the ratio of 3-OH butyric acid to acetoacetic acid, possibly lending support to a diagnosis of a mitochondrial dysfunction (Li et al, 2006; Gutierrez-Quintana et al, 2019).
Imaging findings
Magnetic resonance imaging (MRI) of the brain can reveal, in many of the reported encephalopathic phenotypes in dogs (Table 1), bilateral symmetric lesions at different levels of the brain, typically hypointense on T1-W images, hyperintense on T2-W images, and non-contrast enhancing preferentially affecting grey matter – basal nuclei and brainstem nuclei (Figure 1). Similarly, computed tomography (CT) can demonstrate the same bilateral symmetric lesions with hypodense signal in the regions, typically at the level of the grey matter (Washlag et al, 1999). In leukoencephalomyelopathies, bilateral and symmetrical T2-weighted hyperintensities affecting the brainstem and cerebellar white matter, as well as multifocal T2-weighted hyperintensities in the cerebral white matter, have been reported (Gutierrez-Quintana et al, 2019).
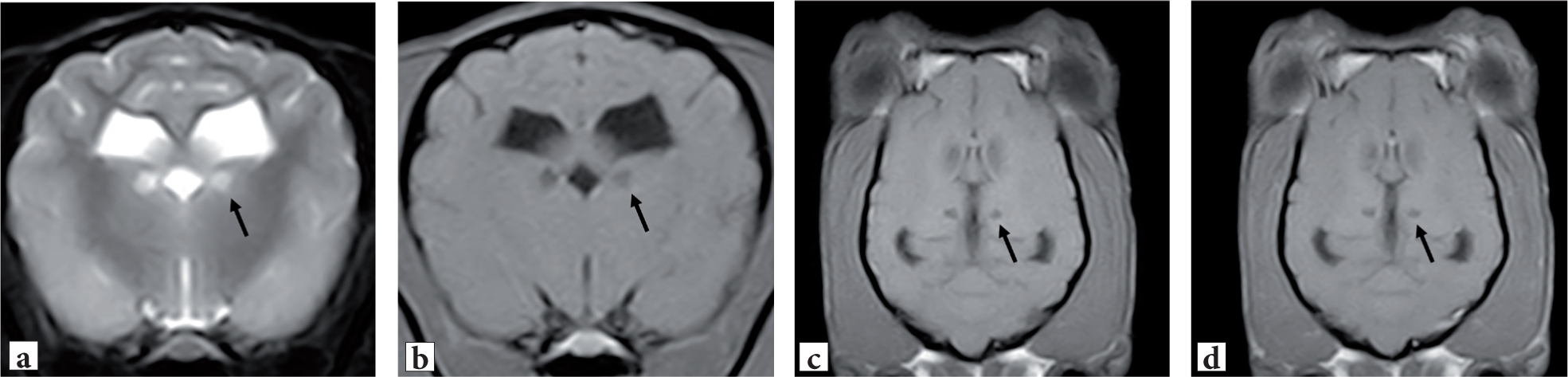
Despite commonly encountered in mitochondrial encephalopathies, basal nuclei malacia is a non-pathognomonic and non-specific finding that may be encountered in other conditions (Barker et al, 2016). Differential diagnosis for bilateral, symmetrical changes within the central nervous system include metabolic diseases (such as hepatic encephalopathy, renal encephalopathy), post-ictal changes (such as following a prolonged status epilepticus), toxicity (such as carbon monoxide), organic acidurias (such as malonic aciduria), lysosomal storage diseases (such as gangliosidosis), nutritional disorders (such as thiamine deficiency) and electrolyte imbalances (such as hypernatremia or salt poisoning).
Histopathological findings
On histopathology, bilateral symmetric, spongy degeneration and/or vacuolar lesions, primarily affecting grey matter, are typically found. Despite being described in several reports and inclusively used to name the disease process, the term spongiform degeneration is considered better reserved for transmissible spongiform encephalopathies (such as scrapie in sheep) with the term spongy degeneration being currently preferred (Vandevelde et al, 2012).
Abnormal mitochondria can be observed on both light and electron microscopy of aff ected neurons and astrocytes. In mitochondrial myopathies, Gomori's trichrome staining can reveal multifocal proliferation and enlargement of mitochondria in tissue sections beneath the sarcolemma, commonly termed ragged red fibres (Vandevelde et al, 2012). In these cases, in vivo muscle biopsies could support diagnosis. Although typically performed in research settings, a mitochondrial enzyme assay can be sought, allowing respiratory chain enzyme activities to be measured in mitochondria from the liver and skeletal muscle (Baiker et al, 2009; Vernau et al, 2013).
Nonetheless, a final diagnosis of mitochondrial encephalopathies, encephalomyelopathies and neuropathies is typically obtained only on post-mortem examination. When post-mortem is performed on cases of a suspected mitochondriopathy, it is important to remember to sample not only the central nervous system but other organs (including the eyes, heart and peripheral musculature), keeping in mind that electronic microscopy or the identification of ‘ragged red fibres’ will typically require fresh-frozen samples (Vandevelde et al, 2012).
Clinical syndromes and classification
Examples of the more extensively studied and recognisable syndromes in dogs include spongiform leukoencephalomyelopathy in the Australian Cattle dog and Shetland Sheepdog, Alaskan Husky encephalopathy in the Alaskan Husky, Leigh-like subacute necrotising encephalopathy in the Yorkshire Terrier and sensory ataxic neuropathy in the Golden Retriever. Clinical phenotypes differ greatly, for example, Alaskan Husky encephalopathy and subacute necrotising encephalopathy resemble Leigh syndrome in children (Brenner et al, 2000; Baiker et al, 2009), affecting young dogs with diffuse progressive signs of predominantly forebrain dysfunction relating mainly to grey matter (such as seizures), whereas spongiform leukoencephalomyelopathy presents more evident signs of white matter dysfunction secondary to hypomyelination (such as generalised tremors). Aside from these more recognisable cases, a mitochondrial mutation has been found in Shetland Sheepdogs with exercise-induced paroxysmal dyskinesia (Nessler et al, 2020), and mitochondrial encephalopathies of unknown cause have also been reported, mostly through individual case reports (Brenner et al, 1997a, 1997 b; Harkin et al, 1999; Gruber et al, 2002; Kent et al, 2009; Collins et al, 2013; Chai et al, 2015). A more detailed description of different neurological phenotypes in canine mitochondriopathies is available in the section outlining reported canine syndromes of confirmed or suspected mitochondrial origin.
A clear classification for mitochondrial diseases in veterinary medicine is lacking. In human medicine, despite the recognition of many clinical phenotypes, any attempts at classification are complicated by genotypes that do not always correlate with the observed phenotype (same mtDNA mutation causing distinct phenotypes), or similar phenotypes correlating to different mutations, adding to the fact that phenotype is also influenced by the level of heteroplasmy (Gorman et al, 2016).
Genetic testing
Mutations in mtDNA were directly incriminated in the development of spongiform leukoencephalomyelopathy in the Australian Cattle Dog and Shetland Sheepdog through a missense mutation in mtDNA encoded cytochrome B (G14474A) (Li et al, 2006), and in sensory ataxic neuropathy in the Golden Retriever through a deletion of a single base pair at position, 5304 in the mitochondrial tRNATyr gene (Baranowska et al, 2009).
Alaskan Husky encephalopathy has been related to a mutation (c.624 insTTGC, c.625 C>A) in the thiamine transporter 2 (SLC19A3) gene, interfering with the uptake of thiamine in the central nervous system via expression of the thiamine transporter protein THTR2. The trait is autosomal recessive, with all clinically-affected dogs homozygous for the trait, and heterozygotes being clinically-normal carriers (Vernau et al, 2013). A mutation in the SLC19A3 gene has also been identified on Yorkshire Terriers with subacute necrotising encephalopathy (Baiker et al, 2009; Drögemüller et al, 2020). All cases reported in one study were homozygous carriers of the mutant allele, an indel variant in exon 2, suggesting that subacute necrotising encephalopathy is also an autosomal recessive disease (Drögemüller et al, 2020).
Recently, paroxysmal exercise-induced dyskinesia in Shetland Sheepdogs has been related to a mutated variant, c.1658G>A or p.Arg553Gln, in the PCK2 gene encoding the mitochondrial phosphoenolpyruvate carboxykinase 2 (Nessler et al, 2020). Although the mitochondrial isoform of this enzyme might be implicated in the aforementioned condition, the authors found no evidence of mitochondrial changes and/or substrate accumulation on histology, enzyme histochemistry, or electron microscopy (Nessler et al, 2020).
Mitochondrial diseases affecting mainly the central and peripheral nervous system in dogs with a published genetic mutation
Alaskan husky encephalopathy/subacute necrotising encephalomyelopathy
Most cases develop clinical signs before 1 year of age. Reported neurological signs include central blindness, proprioceptive positioning deficits, ataxia and seizures, behavioural abnormalities, blindness, facial hypalgesia, tetraparesis and difficulties in prehension of food. Advanced imaging features on MRI of brain include bilateral symmetrical T2-weighted hyperintense, T1-weighted hypointense lesions within the brainstem, extending from the thalamus to the medulla, as well as at the level of the putamen, caudate nucleus and claustrum. Histopathological features include macroscopically identified bilateral and symmetrical cavitated foci within the thalamus, with variable extension into the caudal brain stem. Microscopic lesions include foci of bilateral and symmetrical degeneration in the basal nuclei, midbrain, pons and medulla, as well as multifocal lesions at the base of sulci in the cerebral cortex and in the grey matter of cerebellar folia in the ventral vermis. A genetic mutation has been identified in the thiamine transporter 2 (SLC19A3) gene (Wakshlag et al, 1999; Brenner et al, 2000; Vernau et al, 2013, 2015)
Leigh-like subacute necrotising encephalopathy
The Yorkshire Terrier is affected by Leigh-like subacute necrotising encephalopathy and most cases develop clinical signs between 4 and 18 months, up to 5 years of age. Reported neurological signs include different combinations of disorientation, gait abnormalities, central visual deficits, dysphagia, seizures, altered mentation and muscle fasciculations. Advanced imaging features on CT and MRI of the brain include well circumscribed, non-contiguous bilateral, oblique hypodense (on CT) T2-weighted hyperintense and T1-weighted hypointense (on MRI) lesions within the basal nuclei, mid-thalamus and brain stem, alongside other lesions within the neocortex (Figure 1). Histopathological features include necrotising grey matter lesions with relative preservation of neurons, multiple cerebral infarcts and severe Purkinje-cell degeneration in the cerebellar vermis. A genetic mutation has been identified in the thiamine transporter 2 (SLC19A3) gene (Baiker et al, 2009; Drögemüller et al, 2020).
Inherited spongiform leukoencephalomyelopathy
The Australian Cattle Dog and Shetland Sheepdog are affected by inherited spongiform leukoencephalomyelopathy, and most cases develop clinical signs at 3–4 weeks of age. Reported neurological signs include moderate to severe generalised whole-body tremor and dysmetria, which can progress to more severe ataxia, failure to grow, four limb spasticity and inability to ambulate, with eventual lateral recumbency. In longer-lived individuals, dropped-jaw, hypoglossal dysfunction, nystagmus, dysphagia, and excessive salivation was also identified. Advanced imaging features on CT of the brain included dilation of the lateral and fourth ventricles and diffuse hypomyelination of the white matter (Wood and Patterson, 2001). Histopathological features include widespread vacuolation of myelin in subcortical white matter, cerebellum, brain stem, and spinal cord; occasional scattered demyelinated axons, with some axons appearing to be degenerating, and in severely affected areas, axonal loss and gliosis. A missense mutation in mtDNA encoded cytochrome b (G14474A) was identified (Li et al, 2006).
Sensory ataxic neuropathy
Cases of sensory ataxic neuropathy in Golden retrievers, with an age of onset of 2–8 months of age were reported. Reported neurological signs chronic progressive ataxia, postural reaction deficits, and decreased spinal reflexes with no apparent muscle atrophy, ‘bunny hopping’ at high speed and urinary incontinence. Advanced imaging features of the condition have not been reported. Histopathological features include mild to moderate spinal white matter degeneration with multifocal myelin ballooning, eosinophilic spheroids, intratubar macrophages and fibre loss associated with varying degrees of astroglial proliferation, more pronounced in the fasciculus gracilis and the dorsal part of the lateral funiculus. A deletion in the Mitochondrial tRNATyr Gene has been identified. (Jäderlund et al, 2007; Baranowska et al, 2009).
Paroxysmal exercise-induced dyskinesia
All cases of paroxymal exercise-induced dyskinesia reported to date were female Shetland Sheepdogs with a reported age of onset at 2–6 years of age. Reported neurological signs include episodes of generalised ataxia with hypermetria and muscular hypertonia of all limbs, dystonia, normal to mildly reduced mentation, and a mild tremor with no accompanying autonomic signs. Brain MRI revealed no abnormalities in a single case and no typical histopathology findings have been reported to date. Histopathological findings in muscle and nerve biopsies of a single case have been reported, although these were sparse and featured a diffuse type II fiber predominant muscle atrophy, with no evidence of mitochondrial changes and/or substrate accumulation reported. A mutation was found in the PCK2 gene encoding the mitochondrial phosphoenolpyruvate carboxykinase 2 (Nessler et al, 2020).
Presumed mitochondrial diseases affecting mainly the central and peripheral nervous system in dogs without a published genetic mutation
Spongiform leucoencephalomyelopathy
Cases of spongiform leucoencephalomyelopathy in the Border Terrier, with an age of onset from 3 weeks of age, were reported. Reported neurological signs include cerebellar ataxia and severe generalised coarse body tremors (shaking puppy phenotype), with gradual improvement overtime. Advanced imaging features on brain MRI include bilateral and symmetrical T2-weighted hyperintensities affecting the brainstem and cerebellar white matter and multifocal T2-weighted hyperintensities in the cerebral white matter. Histopathological features include spongiform change affecting the white matter of the cerebellum, brainstem and spinal cord, with decreased myelin content. Gutierrez-Quintana et al (2019) mentioned that a genetic test is available from the University of Missouri, but the gene involved has not yet been reported in the literature (Martin-Vaquero et al, 2012; Gutierrez-Quintana et al, 2019).
Hereditary polioencephalomyelopathy
Cases in Australian Cattle dogs, with an age of onset from 5–24 months, have been reported. Reported neurological signs include seizures, generalised ataxia, and progressive spastic tetraparesis culminating in recumbency. Advanced imaging features on brain MRI include multiple ovoid, bilaterally symmetric lesions T1w hypointense or isointense and T2w hyperintense (malacia of various brain and brain stem nuclei). Spinal cord MRI revealed changes compatible with poliomalacia at the cervical intumescence. Histopathological features include vacuolation of glial cells, dilation of the myelin sheaths and reactive astrocytosis. A genetic mutation has not been reported for this condition. (Brenner et al, 1997b; Harkin et al, 1999).
Mitochondriopathy with regional encephalic mineralisation
Reported as a case report of a 10-month-old female Parson Jack Russell Terrier. Reported neurological signs included progressive ataxia, hypermetria, and deafness. Advanced imaging was not performed. Histopathology investigations revealed severe, bilateral, symmetrical neuronal degeneration and mineralisation of brainstem and cerebellar nuclei. Mineralised deposits were also in myocytes of arteries, hepatocytes and cardiac myocytes, with increased numbers of enlarged or misshapen mitochondria. A genetic mutation was not reported (Gruber et al, 2002).
Encephalomyelopathy with morphological abnormalities in mitochondria
Reported as a case report of a female neutered 16-month-old English Springer Spaniel. Reported neurological signs included ataxia, stumbling into objects and behavioural abnormalities. Advanced imaging was not performed. Histopathology revealed degeneration and astrogliosis of the optic pathways, loss of Purkinje neurons, focal bilateral and symmetrical brain stem spongiosis and diffuse neuroaxial astrogliosis, and giant and bizarre mitochondria within neuronal perikarya and axons. A genetic mutation was not reported (Brenner et al, 1997a).
Polioencephalomyelopathy
Reported as a case report of a 17-month-old Shih Tzu. Reported neurological signs included progressive thoracic limb weakness. Brain MRI revealed a lesion in the spinal cord that extended from the C5 through C7 vertebrae, as well as symmetric lesions in the cranial portion of the cervical spinal cord, caudal colliculi, and vestibular and cerebellar nuclei. Histopathology revealed severe spongiosis of the neuropil with reactive astrocytes (many with high numbers of swollen mitochondria) and preservation of large neurons. A genetic mutation was not reported. (Kent et al, 2009)
Polioencephalomyelopathy resembling Leigh's disease
Reported as a case report of a female neutered 14-month-old crossbreed. Reported neurological signs included exercise intolerance, progressing to non-ambulatory quadriparesis and severe dyspnoea. Advanced imaging was not performed. Histopathological findings include bilateral and symmetrical malacia/cavitation affecting grey matter in the medulla oblongata (part of the olivary nuclei) and caudal cervical and cranial thoracic spinal cord. A genetic mutation was not reported (Chai et al, 2015).
Subacute necrotising encephalopathy
Cases in American Staffordshire Bull Terriers, with an age of onset between 6–8 weeks, were reported. Reported neurological signs included rapidly progressive ataxia, head tilt, head bobbing, nystagmus and strabismus. Advanced imaging features of the condition have not been reported. Histopathological features include malacic lesions at level of vestibular nuclei, solitary tract and nucleus, olivary nucleus and the medial vestibular nucleus. Evidence of bilaterally symmetrical necrotising encephalopathy, extensive neovascularisation, gliosis of vestibular nuclei, preservation of neuronal cell bodies, alongside malacia and vacuolation of the neuropil. A genetic mutation has not been reported for this condition (Collins et al, 2013).
Mitochondrial myopathy in the dog
Mitochondrial myopathy relating to a pyruvate dehydrogenase deficiency (PDP1) was reported on the Clumber Spaniel and Sussex Spaniel (Cameron et al, 2007). Other mitochondrial myopathies were reported in the German Shepherd Dog (Paciello et al, 2003), Springer Spaniel (Tauro et al, 2008), Jack Russel Terrier (Olby et al, 1997), Old English Sheepdog (Breitschwerdt et al, 1992; Vijayasarathy et al, 1994), Irish Terrier (Wentnik et al, 2010), however a genetic mutation has not been yet identified in these breeds. (Griffiths and Duncan, 1979; Herrtage and Houlton, 1979; Houlton and Herrtage, 1980; Shelton et al, 2000; Abramson et al, 2004; Cameron et al, 2007).
Treatment and outcome
Currently, there is no therapy that has been proven to slow down or prevent the progression of mitochondrial encephalopathies in dogs, and most cases will invariably be euthanised within weeks to months of the onset of neurological dysfunction. Palliative care, for instance institution of anti-epileptic medication when seizures are present, may be necessary in some cases. In cases of disorders affecting the Krebs cycle, a ketogenic diet might be beneficial to improving mitochondrial function and decreasing oxidative stress (Wakshlag et al, 1999). Limited therapeutic modalities, such as diet modification (gluten-free diet), stress reduction or institution of acetazolamide or zonisamide (in a single case), have led to improvement in Shetland Sheepdogs presenting with exercise-induced paroxysmal dyskinesia (Nessler et al, 2020).
In humans, therapy for mitochondrial diseases is mostly palliative with nutritional supplementation (such as ubiquinone – coenzyme Q or analogues). Avenues of therapeutic research in human mitochondriopathies include the elimination of accumulative noxious compounds (such as lactate), shifting of heteroplasmy (lowering the mutated mDNA load within a cell), alteration of mitochondrial dynamics (trying to eliminate dysfunctional mitochondria, or potentiating normally functioning mitochondria), gene therapy and cytoplasmic transfer (for example, by means of producing an embryo with nDNA of the biological parents but mtDNA of a non-affected donor) (Di Mauro et al, 2013).
Conclusions
A clear classification for mitochondrial diseases in veterinary medicine is lacking, and this review is the first to specifically address mitochondrial encephalopathies, encephalomyelopathies and neuropathies in dogs. Owing to the varied clinical phenotypes of mitochondrial diseases, the distinct aetiologies of mitochondrial dysfunction (such as inherited mutations in mtDNA or nDNA coding for mitochondrial components) and the phenomenon of heteroplasmy within the individual, a definitive classification in dogs will be difficult to achieve. As recognition of mitochondrial disorders in dogs increases, further clinical phenotypes and specific genetic mutations are expected to be found and patterns described, allowing the classification of mitochondriopathies into more recognisable syndromes, as is currently the case in human medicine.
KEY POINTS
- Mitochondriopathies are considered rare in the dog.
- Mitochondriopathies occur most typically in young dogs with progressive signs of multifocal neurological dysfunction.
- Evidence in serum biochemistry or cerebrospinal fluid of increased lactate and/or lactate:pyruvate concentrations could raise suspicion of a mitochondriopathy.
- The presence of bilaterally symmetrical lesions, particularly affecting grey matter on both brain or spinal cord magnetic resonance imaging and computed tomography, could indicate a nutritional/metabolic disorder, but also a mitochondriopathy.
- Clinical phenotypes are varied and only a few canine mitochondriopathies have been described with known specific genetic mutations.
- Therapeutic dietary modalities have been to some benefit in a handful of cases, but overall prognosis is poor.
- Increased recognition of mitochondrial disorders is expected to allow future classification into recognisable syndromes, as is currently the case in human medicine.