


Ventricular arrhythmias are abnormal heart rhythms that originate in the ventricles. They can range from isolated and relatively benign to complex and life threatening. The mechanisms of these are complex and can be difficult to determine based on a surface electrocar-diogram, but analysis can guide differential diagnoses and anti-ar-rhythmic drug selection. Juvenile inherited arrhythmias are documented in the literature and particularly investigated in German Shepherd dogs, with sporadic reports of inherited arrhythmias in other breeds (Moïse et al, 1994; Reuter et al, 2024). This case report describes the approach to diagnosis and treatment of a complex ventricular arrhythmia initially documented in a 3-month-old asymptomatic Dachshund.
A 3-month-old female Dachshund was referred following detection of an arrhythmia by the attending veterinarian at a routine vaccination appointment. There were no reported clinical signs. Physical examination revealed an alert and ambulatory patient, weighing 4.1 kg with reduced body (World Small Animal Veterinary Association score – 3/9; World Small Animal Veterinary Association, 2013a) and muscle condition (World Small Animal Veterinary Association score – B; World Small Animal Veterinary Association, 2013b). The patient's heart rate was 148 bpm with an irregularly irregular rhythm and pulse deficits. There was no audible heart murmur. Systolic blood pressure (Doppler method) was 130 mmHg.
An electrocardiogram (Figure 1) revealed an underlying sinus rhythm with frequent wide and aberrant complexes, negative in leads I, II, III and aVF, most consistent with ventricular ectopy of left ventricular origin. These occurred as singles, couplets, triplets and runs, interspersed with sinus rhythm and occasional fusion complexes (Figure 1). The heart rate was 170 bpm with an irregularly irregular R–R interval that tended to accelerate during a ventricular paroxysm (shortening R–R interval). The sinus P–QRS–T complexes were all within reference ranges for amplitudes and durations. The QT interval was within reference at 180 ms (reference range: 150–240 ms) and within reference when corrected for R–R using the logarithmic formula (QTc=log 600×QT/log RR) 200 ms (reference range: 150–240 ms) (Matsunaga et al, 1997). There were examples of a QT/TQ ratio >1, which has been associated with increased risk of arrhythmia (Fossa, 2008) (Figure 1). Underlying sinus rhythm with frequent and complex ventricular ectopy was the prominent differential, with a supraventricular localisation (with aberrant conduction) excluded by the normal sinus QRS complex morphology and presence of fusion complexes. Enhanced automaticity was considered a possible mechanism of arrhythmia precipitation, based on the progressively shortening R–R interval consistent with a ‘warming up’ foci. The fastest instantaneous ventricular rate was 280 bpm, though no R on T phenomenon was observed on the six-lead trace. The coupling interval between the preceding sinus complex and first ventricular ectopic was often, but not exclusively, consistent (~300 ms).
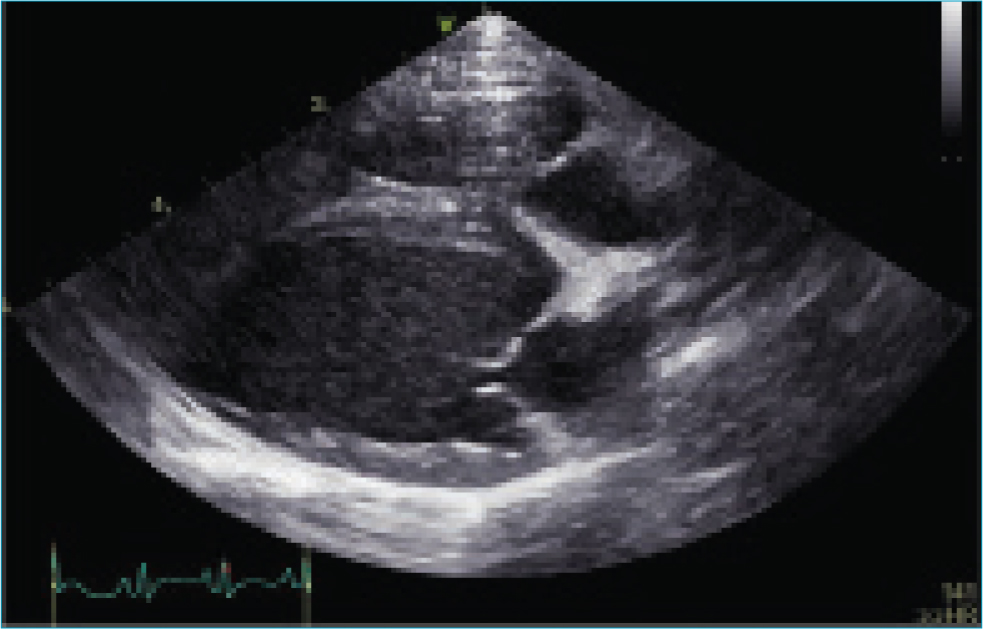
Doppler echocardiography identified a structurally and functionally normal heart (Figure 2). The left atrium was normal in size (short axis diastolic left atrium/aorta 1.33; reference range: <1.6) (Hansson et al, 2002). The left ventricular internal diameters (LVID) were within reference ranges based on normalisation for body weight (n) by allometric scaling, in both diastole (D) and systole (S) (LVIDDn 1.49, reference range <1.7; LVIDSn 0.96 <1.0, reference range 0.55–0.96) (Visser et al, 2019). Systolic function was adequate based on Simpson's derived ejection fraction (70%; reference range: <50%) and end systolic volume indexed for body weight (0.77 ml/kg; reference range: <1.5) (Visser et al, 2019). The myocardium echotexture appeared unremarkable. There was trivial mitral and tricuspid regurgitant flow, occurring predominantly during ventricular contractions triggered by the ectopic depolarisations. The valves were morphologically normal, and this insufficiency was suspected to reflect loss of systolic coordination or synchrony.
Cardiac troponin I (cTnI – IMMULITE assay) was within laboratory reference (0.032 ng/mL; reference range: <0.15), not consistent with recent or ongoing myocardial cell damage. Serum biochemistry analysis identified non-specific changes consistent with age:
Haematology identified a mild thrombocytosis (470x109/L; reference range: 150–400x109/L) with no further abnormalities.
A 24-hour ambulatory electrocardiographic monitor was fitted and the patient was discharged. There were no adverse events reported during the monitoring period. The predominant rhythm was sinus rhythm interspersed with frequent wide, bizarre QRS complexes without P-QRS coupling, assumed to be ventricular complexes that occurred singularly and as multiple consecutive beats. There were 92 895 of these over the recording period. Approximately 20% of these were single ventricular ectopic complexes. There were examples of bigeminal and trigeminal patterns. There was significant complexity with couplets, triplets, runs and ventricular tachycardia with examples of R on T phenomenon. During ventricular tachycardia, there were examples of acceleration with progressive shortening of consecutive R–R intervals as identified on the electrocardiogram, but this was not always present (Figures 3 and 4). There was a trend toward an increased number and complexity of ventricular arrhythmias in the early hours of the morning, presumably during sleep (Figures 5 and 6). The coupling interval between the preceding sinus complex and first ventricular ectopic was usually fairly consistent at 300 ms.
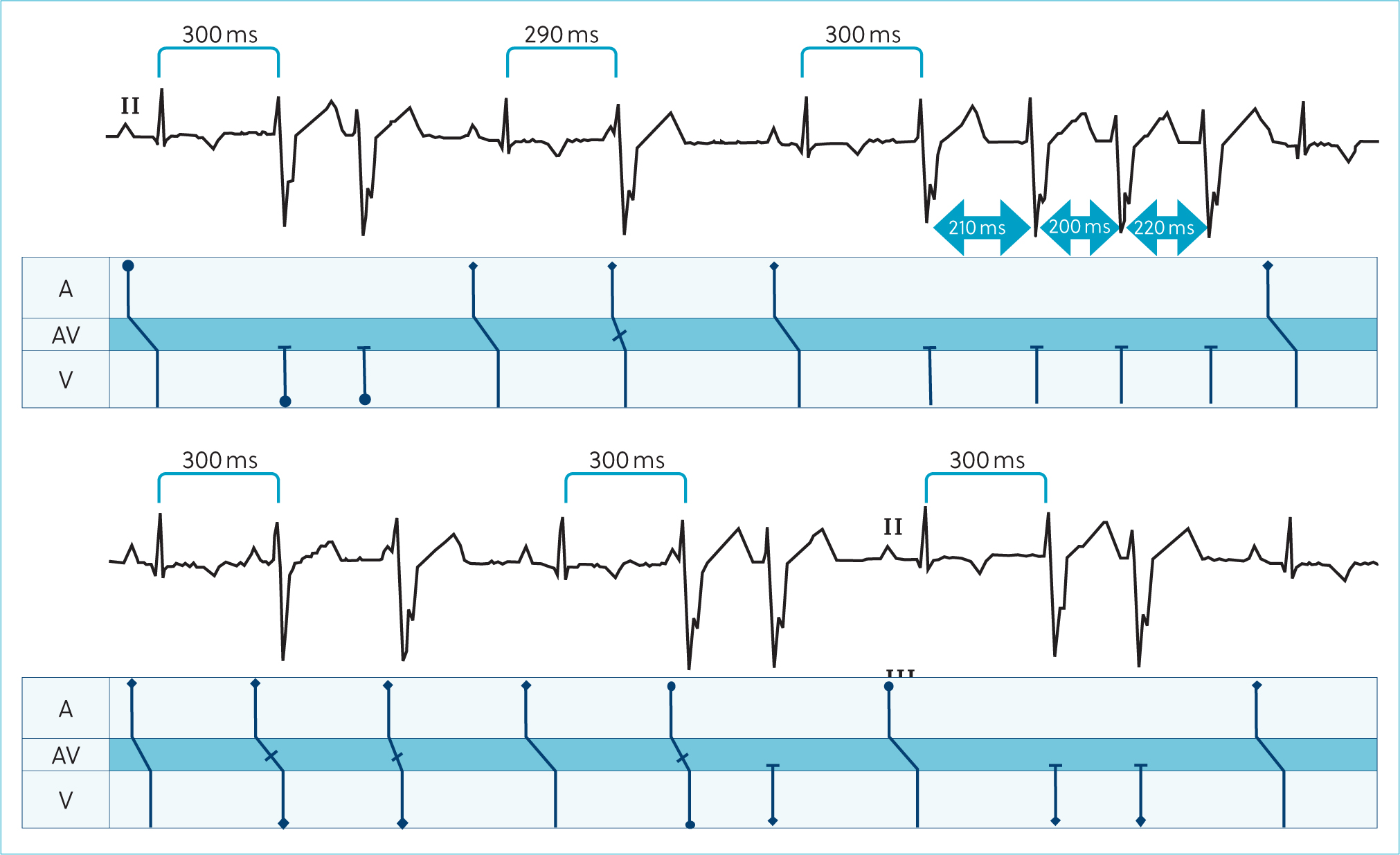
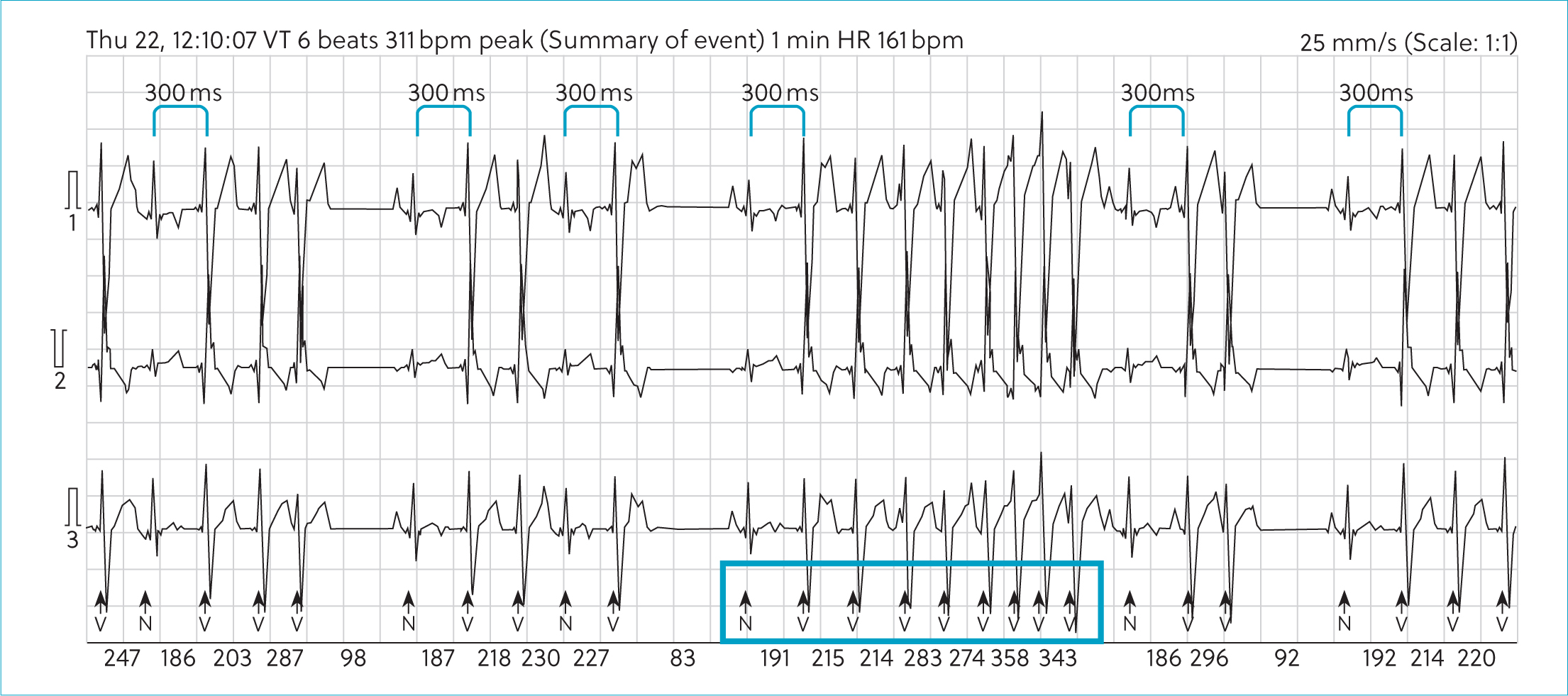
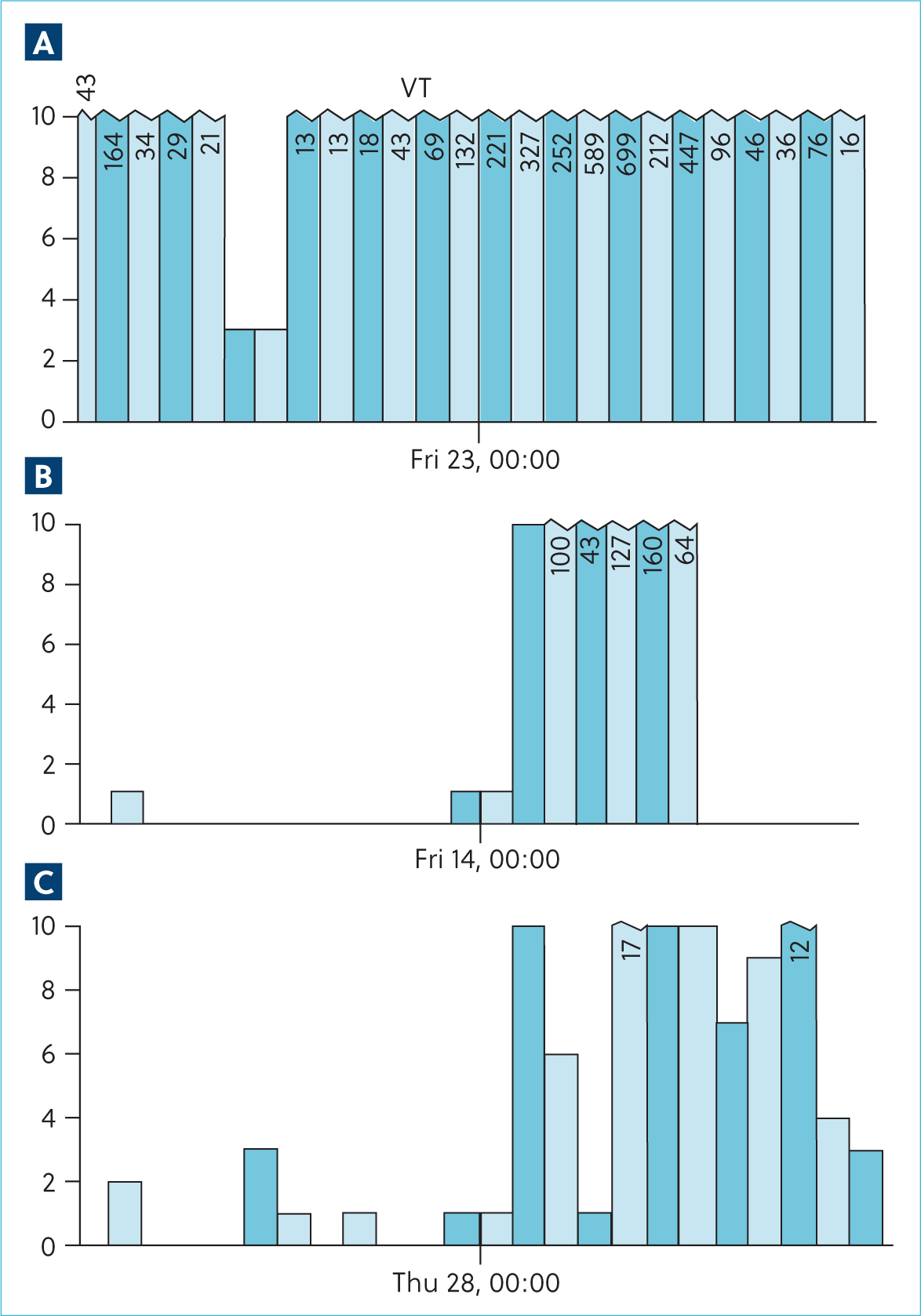
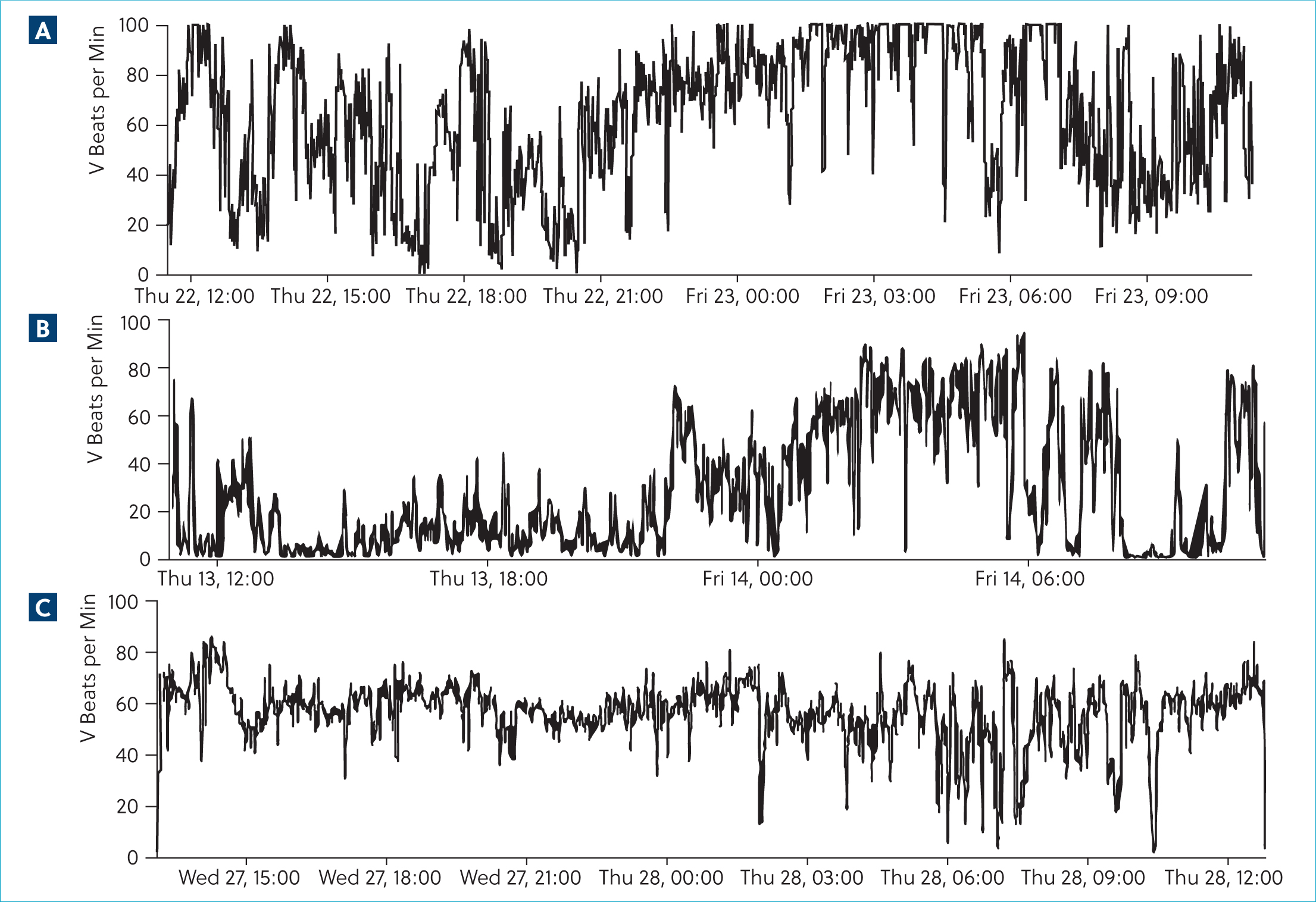
Oral anti-arrhythmic therapy was started with oral sotalol (So-talol Hydrochloride, Tillomed) (2.4 mg/kg) every 12 hours and oral mexiletine (Mexiletine Hydrochloride, BOVA) (6 mg/kg) every 8 hours. A 24-hour ambulatory electrocardiogram (Holter) was repeated after 14 days, during which time the patient was reported to be active at home. On reassessment, physical examination was unchanged, but her body weight had increased to 5.9 kg. Repeat ambulatory electrocardiographic monitoring identified a trend towards a decrease in ventricular ectopy number and complexity. There was a 55.4% reduction in the number of ventricular aberrant complexes; however, day-to-day variability of up to 80% has been reported in Boxers with arrhythmogenic right ventricular cardiomyopathy (Spier and Meurs, 2004). There was also a reduction in complexity with a reduction in the incidence of ventricular tachychardia. Again, there was a predilection for increased ventricular arrhythmias during sleeping hours (Figures 5 and 6).
Oral magnesium supplementation (Magnesium citrate) was added to the treatment protocol (250 mg once daily, ~42 mg/kg). At last contact, 2 months following diagnosis, the patient remained clinically unaffected, but repeat ambulatory electrocar-diographic monitoring demonstrated that the incidence of ventricular arrhythmias had increased by 48.6%, only 13.2% fewer than the first assessment. Complexity was broadly similar; however, there was a further reduction in the incidence of ventricular tachycardia, from 507 during the second Holter to 98 following the addition of magnesium. The tendency towards ventricular arrhythmia increasing during sleeping hours was not sustained in this recording, complicating interpretation of the underlying mechanism.
Discussion
This case report describes the approach to, and medical treatment of, complex ventricular arrhythmias in a young dog. Ventricular arrhythmias can present as an incidental finding, as in this case; however, they can result in significant morbidity including syncope and exercise intolerance, as well as potentially increasing the risk of sudden death.
Ventricular arrhythmias originate in the ventricular myocardium and depolarisation does not involve the Purkinje system but propagates in a slow ‘cell-to-cell’ manner resulting in the typical wide appearance of the QRS complex on the surface electrocar-diogram, and opposite polarity of the T wave since repolarisation follows a similar cell to cell pathway (Willis, 2018). Wide QRS complexes of ventricular origin must be differentiated from supraventricular complexes conducted with aberrancy as a result of damage (such as a bundle branch block) or rate-related impaired conduction through the Purkinje system (Santilli et al, 2012). This was unlikely in the present case because of the presence of narrow QRS complexes identified during sinus complexes. The presence of fusion complexes also provides evidence of a ventricular ectopic focus since the ectopic focus and sinus complexes depolarising through the ventricular myocardium merge. Ventricular arrhythmias are not a primary diagnosis and suggest an underlying pathology resulting in mechanisms that precipitate arrhythmias.
Attempts to characterise the mechanism of an arrhythmia based on a surface electrocardiogram can be challenging, but it is crucial to better understand the underlying aetiology (Box 1). Ventricular arrhythmias may be caused, and sustained, by enhanced automaticity, triggered activity, re-entry or a combination of these mechanisms (Willis, 2018). Enhanced automaticity describes a situation where the discharge rate of cardiac pacemaker cells becomes abnormally high; in ventricular arrhythmias, these pacemaker cells would be within the ventricular myocardium. This may occur because of a reduction in threshold potential or a higher resting membrane potential, resulting in an increased likelihood of spontaneous discharge (Gaztañaga et al, 2012). It can also be caused by sympathetic stimulation or hypokalaemia and is the mechanism resulting in idioventricular rhythms often displaying gradual acceleration and deceleration (Ilvento et al, 1982).
Electrocardiographic features of different arrhythmia mechanisms
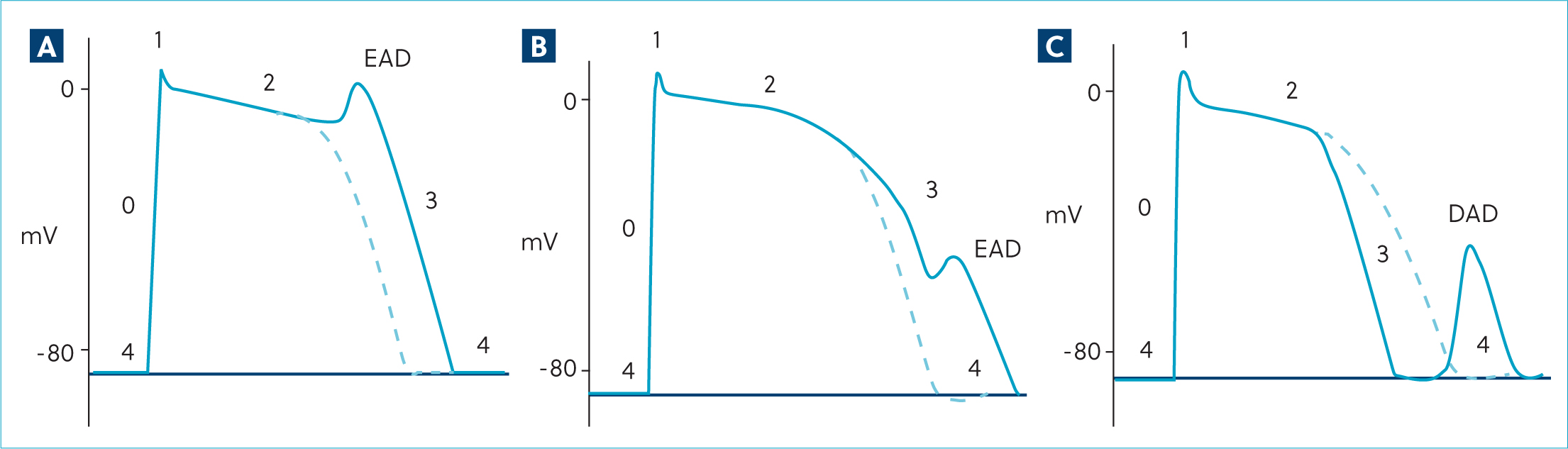
Ventricular arrhythmias caused by triggered activity result from premature depolarisations before full repolarisation, described as early after depolarisations or, if occurring during phase four of the action potential before the next sinus-initiated complex, as delayed after depolarisations (Figure 7) (Gaztañaga et al, 2012). Early after depolarisations are caused by factors that increase the action potential duration or cause more positive membrane potential during stage two (plateau) and three (repolarisation) of the action potential. Usually, reopening of inward calcium currents result in stage two early after depolarisations (Yamada et al, 2008), and fluctuations in the potassium current are implicated in stage three early after depolarisations (Maruyama et al, 2011). Slower heart rates (eg during sleep) will lengthen the action potential duration, prolonging the time in this vulnerable period. Early after depolarisations may be the cause of ventricular arrhythmias in long QT syndrome, either as a result of a channelopathy or induced pharmacologically (eg by procainamide, a class 1A antiarrhythmic drug) or sotalol/amiodar-one (class III anti-arrhythmic drugs) (Kannankeril et al, 2010). Generally, these early after depolarisations are more prevalent at slower heart rates. Delayed after depolarisations occur during stage four (resting state) and are caused by excessive intracellular Ca2+ levels such as during ischaemia, secondary to defects in calcium cycling in heart failure, or drug side effects such as digoxin toxicity. Excessive intracellular Ca2+ drives inward movement of Na+ via cell membrane 3Na+/1Ca2+ exchangers, resulting in premature depolarisations. In contrast to early after depolarisations, the rate and amplitude of delayed after depolarisations increase with shorter R–R intervals (ie at faster heart rates) (Gaztañaga et al, 2012).
Re-entry occurs when myocardial cells regain excitability before full depolarisation, allowing propagation of a wavefront around less excitable, still refractory cells. A unidirectional block is required in one arm with slowed conduction in the other, so the circuit can be perpetuated by a constantly excitable leading edge (Kléber and Rudy, 2004). This is usually caused by disease affecting the myocardium resulting in a population of cells with heterogenous excitability. Re-entrant arrhythmias are typically triggered by a premature beat and are sudden in onset and offset and tend to have a regular R–R interval during runs (ie no warm up or cool down) (Kléber and Rudy, 2004).
Acute management of ventricular arrhythmias with an intravenous anti-arrhythmic may be indicated in the first instance if the patient is haemodynamically unstable. Generally, this is only required with persistent rapid ventricular tachycardia (heart rate>180–200 bpm) (Figure 8). Once stable, diagnostics must focus on investigation of a primary disease process. Systemic disease is an important cause of ventricular arrhythmias (Kittleson, 1998). Anaemia, abdominal neoplasia, trauma, systemic inflammatory response syndrome and hypokalaemia are important examples that must be quickly ruled in or out to allow targeted therapy (Keyes et al, 1993). In this case, the patient's signalment and history did not suggest systemic disease and haematology and serum biochemistry did not identify clinically significant changes. Abdominal imaging was indicated in this case and considered essential in an older dog but was not prioritised because of financial considerations. Geographical location and recent history should also be a consideration given the reports of ventricular arrhythmias associated with European adder bites (Harjen et al, 2020).

Structural cardiac disease is another important cause of ventricular arrhythmias and requires echocardiography to diagnose. Clinically significant ventricular arrhythmias are less common in myxomatous mitral valve disease and congenital heart disease than in patients with myocardial disease (Rasmussen et al, 2012). Complex ventricular ectopy is a key feature of canine cardiomyopathy, and patient signalment (breed, age of onset) can help to increase the index of suspicion, eg Dobermanns and Great Danes with dilated cardiomyopathy and Boxer dogs or English bull dogs with arrhythmogenic right ventricular cardiomyopathy (Wess, 2022). Normal echocardiography does not exclude the arrhythmic forms of either of these conditions in an individual, and an ambulatory electrocardiogram is indicated to detect cases that have arrhythmias with no structural disease (Dukes-McEwan et al, 2022). Infective endocarditis and myocarditis are also important differentials in cases with ventricular arrhythmias. In the present case, these were considered less likely based on normal echocardiography and a normal cTnI (Kilkenny et al, 2021). Specific testing can be performed for commonly implicated pathogens in myocarditis such as Borrelia, Bartonella, parvovirus and toxoplasma.
In this case, diagnostic investigations did not identify a significant underlying cardiac or systemic disease as would be expected in a juvenile dog with ventricular arrhythmias. In humans, genetic mutations resulting in ion channelopathies are implicated in the development of ventricular arrhythmias in affected individuals (Offerhaus et al, 2020). These are also reported in a few specific breeds of dog (Box 2). There are no reports of inherited channelopathies in Dachshunds in the veterinary literature.
Summary of reported familial and inherited ventricular arrhythmias
The most extensively studied familial ventricular arrhythmia in dogs is in the German shepherd dog; however, specific mutations have not yet been identified (Moïse et al, 1994). This disease affects young German shepherds, and affected individuals have increasing ventricular arrhythmias and risk of death within the first year of life followed by regression of the ventricular arrhythmias after 2 years of age (Moïse et al, 1997a). These ventricular arrhythmias are distinctive in that they are pause-dependent, occurring during periods of sinus bradycardia, and frequently occur as rapid polymorphic ventricular tachycardia (Moïse et al, 1997b). Detection may not be possible on routine assessment, and the first clinical manifestation may be sudden death during sleep or ventricular arrhythmias may be noted when patients are anaesthetised for neutering. The mechanism of the arrhythmia has been identified as early after depolarisations originating in the Purkinje fibres of the left ventricle (Moïse et al, 1997a; 1997b). Repolarisation is not homogenous across the left ventricle, particularly at longer cycle lengths, which is attributed to differences in transmembrane potassium currents and calcium cycling (Gilmour and Moïse, 1996; Jesty et al, 2013). Sympathetic denervation of the myocardium has also been implicated (Dae et al, 1997). It has been shown that in these individuals the combination of oral mexiletine (8 mg/kg three times daily) and oral sotalol (2.5 mg/kg twice daily) was most effective at reducing arrhythmia frequency than monotherapy of either drug (Gelzer et al, 2010). Additionally, sotalol monotherapy exacerbates ventricular arrhythmias by prolonging the action potential and predisposing to early after depolarisations (Gelzer et al, 2010).
Familial ventricular arrhythmia has also been reported in the English Springer Spaniel, Rhodesian Ridgeback and Leonberger breeds. QT interval prolongation associated with a missense mutation in the KCNQ1 gene has been reported in a family of English Springer Spaniels, resulting in a high incidence of sudden death during activity in affected dogs (Ware et al, 2015). This mutation was thought to be similar to that reported in people with long QT syndrome subtype 1 (LQT1), with increased risk reported during exercise. In the Rhodesian Ridgeback, a familial cardiac arrhythmia has been identified with a juvenile age of onset and an autosomal recessive pattern of inheritance (Meurs et al, 2016). This was associated with a single missense variant in the QIL1 gene, producing mitochondrial cristae abnormalities resulting in ventricular arrhythmias (Meurs et al, 2019). Ventricular arrhythmias have also been described in the Leonberger, associated with sudden death in individuals less than 3 years of age with no structural cardiac disease identified during echocardiography (Wiberg et al, 2020). In this breed, death typically occurred during sleep or rest shortly after exercise. In this cohort, the QT interval was not different to that of unaffected Leonbergers. The mode of inheritance and genetic association have not been published at this time, but an inherited channelopathy is suspected in this breed.
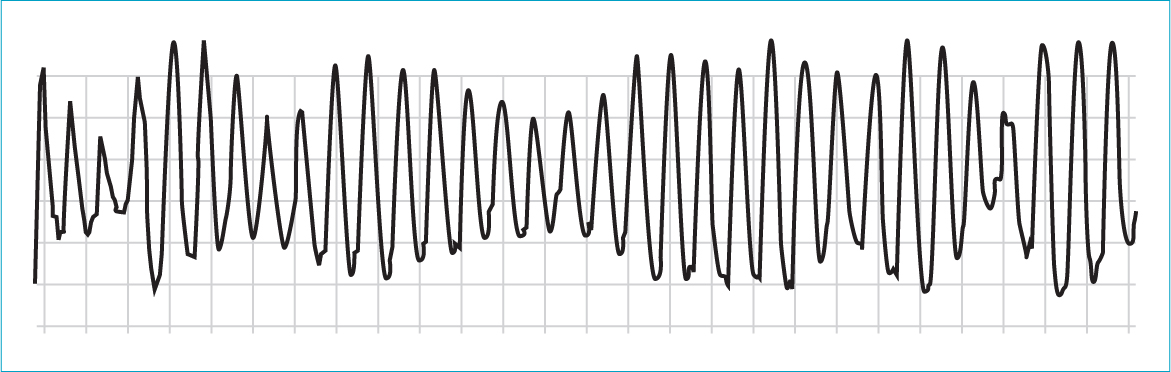
Inherited ventricular arrhythmias associated with channelopathies have been extensively described in humans (Box 3). These are a group of inherited genetic mutations with different phenotypes and incomplete penetrance that predispose individuals to ventricular arrhythmias and an increased risk of sudden death. They are diagnosed based on characteristic clinical history, genetic testing and electrocardiographic changes which may only be revealed by exercise or pharmacological challenge. Long QT syndrome is the most common of these, affecting one in 2500 individuals, with 5% of affected individuals suffering a fatal arrhythmia as the first event (Behere and Weindling, 2015). Individuals are at risk of the unstable polymorphic ventricular tachycardia called torsades de pointes responsible for symptoms (syncope, palpitations) and torsades de pointes may degenerate to ventricular fibrillation (Figure 9). Multiple subtypes are reported with differing triggers for torsades de pointes, including exercise (LQT1), sudden stressful events (LQT2) and sleeping (LQT3).
Summary of inherited arrhythmias reported in humans (Offerhaus et al, 2020)
Treatment of ventricular arrhythmia centres on manipulation of the myocardial action potential, but it is important to consider that anti-arrhythmic treatment can also precipitate or exacerbate arrhythmias and must be considered carefully. Although the aim of therapy is to reduce the incidence and complexity of ventricular arrhythmias, there is no strong evidence that this reduces an individual's risk of sudden death. In this case, the authors' elected to treat with the combination of mexiletine (Class IB) and sotalol (Class III) as described in German shepherd dogs (Gelzer et al, 2010). Class I anti-arrhythmic drugs are sodium channel blockers that slow the rapid phase 0 depolarisation of the action potential, ultimately slowing the speed of cell-to-cell conduction. The class IB anti-arrhythmic drugs, such as lidocaine or mexiletine, slightly shorten the action potential duration, whereas class IA agents will lengthen it. Sotalol is a class III antiarrhythmic, blocking K+ channels involved in repolarisation, which leads to prolongation of the action potential. It also has non-selective beta-blocker effects. The combination of mexiletine and sotalol may be synergistic and superior in controlling ventricular arrhythmias caused by triggered activity, particularly early after depolarisations (Gelzer et al, 2010).
The addition of magnesium was instigated on the suspicion that the ventricular arrhythmias were pause-dependent. Magnesium shortens the action potential duration and can be protective against early after depolarisations (Bailie et al, 1988). Serum magnesium was normal on biochemistry (0.832 mmol/L; reference range: 0.7–1.0 mmol/L), but as an intracellular ion, serum levels may not reflect intracellular concentrations.
Based on the wide day-to-day variability in incidence and complexity of ventricular arrhythmias reported, the changes in the number and complexity of ventricular arrhythmias over time did not reach a threshold that convincingly demonstrated successful therapeutic intervention in this case. However, the trend towards reduced incidence of ventricular tachycardia may be relevant (Figure 5). Additionally, the most recent ambulatory electrocardiogram did not demonstrate any pause-dependence, and therefore, does not provide further support for early after depolarisations as the underlying mechanism. Whether this represents variability, differing mechanisms over time or the impact of therapy is unknown.
Conclusions
This case describes the approach to, and management of, ventricular arrhythmias in a juvenile Dachshund. Based on signalment, electrocardiographic findings and exclusion of other possible differentials, a genetic mutation resulting in a channelopathy was suspected as the cause of this arrhythmia. This diagnosis cannot be definitive without genetic testing in a larger cohort of affected dogs, and to the authors knowledge, there are no other reports of juvenile ventricular arrhythmias in this breed.